عمومية
الورم الأرومي الشبكي (Rb) هو ورم خبيث في العين ينشأ من خلايا شبكية العين. يمكن أن يحدث هذا السرطان في أي عمر ، ولكن ظهوره يكون أكثر شيوعًا خلال فترة الرضاعة قبل سن الخامسة.

يعتبر سرطان الأطفال عدوانيًا: يمكن أن ينتشر الورم الأرومي الشبكي إلى الغدد الليمفاوية أو العظام أو نخاع العظام. نادرًا ما يصيب الجهاز العصبي المركزي (الدماغ والحبل الشوكي).
حوالي 90٪ من الأطفال المصابين بالورم الأرومي الشبكي لديهم تشخيص إيجابي (احتمال الشفاء) ، بشرط أن يكون التشخيص مبكرًا وأن يبدأ العلاج قبل انتشار السرطان. كلما كان ذلك ممكنًا ، يكون الهدف من التدخل الطبي هو الحفاظ على رؤية المريض.
الأسباب
سلسلة الأحداث التي تؤدي إلى ظهور الورم معقدة ، تبدأ عندما تطور الخلايا في شبكية العين طفرة (أو حذف) ، بما في ذلك الجين الكابت للورم RB1 ، والموجود على النطاق q14 من الكروموسوم 13 (13q14).
تحتوي كل خلية عادة على جينين RB1:
- إذا كانت هناك نسخة واحدة على الأقل من الجين تعمل بشكل صحيح ، فلا ينشأ الورم الأرومي الشبكي (لكن الخطر يزيد) ؛
- عندما يتم تحوير نسختين الجين أو فقدهما ، يحدث تكاثر الخلايا غير المنضبط.
في كثير من الحالات ، ليس من الواضح ما الذي يحث بالضبط على التغييرات في جين RB1 (ورم أرومي شبكي متقطع) ؛ يمكن أن تنتج عن أخطاء وراثية عشوائية تحدث ، على سبيل المثال ، أثناء التكاثر والانقسام الخلوي. ومع ذلك ، فمن المعروف أن التشوهات الجينية الكامنة وراء الورم الأرومي الشبكي يمكن أيضًا أن تنتقل من الآباء إلى الأطفال ، بنمط وراثي سائد وراثي. هذا يعني أنه إذا كان أحد الوالدين يحمل جينًا متحورًا (سائدًا) ، فسيكون لكل طفل فرصة بنسبة 50٪ في أن يرثه ، وفرصة بنسبة 50٪ في الحصول على تركيبة جينية طبيعية (جينات متنحية).
- تعمل خلية عرضية على تعطيل نسختها الطبيعية الوحيدة من جين RB1 (نسخة واحدة تم تحورها بالفعل) ؛
- يؤدي فقدان نسختين من RB1 إلى "تكاثر مفرط لشبكية العين.
- تقوم خلية عرضية بتعطيل أحد جيناتها الطبيعية RB1 ؛
- النسخة الثانية من جين RB1 معطلة ؛
- يؤدي فقدان نسختين من RB1 إلى تكاثر مفرط للخلايا يؤدي إلى الورم الأرومي الشبكي.
الخصائص الوراثية والجزيئية
- كان الورم الأرومي الشبكي أول ورم يرتبط ارتباطًا مباشرًا "بشذوذ جيني (حذف أو طفرة في مجموعة q14 من الكروموسوم 13).
- يقوم RB1 بترميز بروتين pRb ، الذي يلعب دورًا رئيسيًا في دورة الخلية: فهو يسمح بتكرار الحمض النووي وتطور دورة الخلية ، حيث يشارك في التحكم في نسخ جينات المرحلة S (G1 → † "S).
- بالإضافة إلى الورم الأرومي الشبكي ، يتم تعطيل جين RB1 في سرطانات المثانة والثدي والرئة.
ورم أرومي شبكي وراثي
يميل الأطفال المصابون بالورم الأرومي الشبكي الوراثي إلى الإصابة بالمرض في سن مبكرة أكثر من الحالات المتفرقة. علاوة على ذلك ، فإن هؤلاء الأطفال معرضون لخطر متزايد للإصابة بسرطانات غير عينية أخرى ، لأن الشذوذ في جين RB1 خلقي (أي موجود منذ الولادة) ويؤثر على جميع خلايا الجسم (المعروفة باسم طفرة السلالة الجرثومية) ، بما في ذلك كلاهما. شبكية العين: لهذا السبب ، غالبًا ما يعاني الأطفال المصابون بالشكل الوراثي من ورم أرومي شبكي ثنائي بدلاً من عين واحدة فقط.
أعراض
لمعرفة المزيد: أعراض الورم الأرومي الشبكي
العلامة الأكثر شيوعًا ووضوحًا للورم الأرومي الشبكي هي المظهر غير الطبيعي للبؤبؤ ، والذي يظهر انعكاسًا أبيض مائلًا للرمادي عندما يصطدم بشعاع من الضوء (ابيضاض الدم أو منعكس القط غير الطبيعي). تشمل العلامات والأعراض الأخرى: انخفاض الرؤية ، وألم العين واحمرارها ، وتأخر النمو. قد يصاب بعض الأطفال المصابين بالورم الأرومي الشبكي بحول (عيون منحرفة) ؛ في حالات أخرى ، من الممكن العثور على الجلوكوما الأوعية الدموية الجديدة ، والتي يمكن أن تسبب بعد فترة من الوقت تضخم العين (buftalmo).
يمكن للخلايا السرطانية أن تغزو العين والبنى الأخرى بشكل أكبر:
- ورم أرومي شبكي داخل العين. يمكن تعريف الورم الأرومي الشبكي على أنه داخل مقلة العين عندما يكون الورم موجودًا بالكامل داخل العين. يمكن العثور على الورم فقط في شبكية العين أو يؤثر أيضًا على أجزاء أخرى ، مثل المشيمية والجسم الهدبي وجزء من العصب البصري. لذلك ، لا ينتشر الورم الأرومي الشبكي داخل العين إلى الأنسجة حول الجزء الخارجي من العين.
- ورم أرومي شبكي خارج العين. يمكن أن يتكاثر الورم ليؤثر على الأنسجة حول العين (الورم الأرومي الشبكي المداري). يمكن أن ينتشر السرطان أيضًا إلى مناطق أخرى من الجسم ، مثل الدماغ والعمود الفقري ونخاع العظام والعقد الليمفاوية (الورم الأرومي الشبكي النقيلي).
من المعروف أن وجود التمدد المداري ، والتورط العنبي ، وغزو العصب البصري من عوامل الخطر لتطوير ورم أرومي الشبكي النقيلي.
تشخبص
في حالة وجود تاريخ عائلي إيجابي ، يخضع المريض لفحوصات عين منتظمة لفحص السرطان. إذا كان الورم الأرومي الشبكي الخلقي ثنائيًا ، فعادة ما يتم تشخيصه في السنة الأولى من العمر ، بينما عندما يصيب عينًا واحدة فقط ، يمكن تأكيد وجود الورم في حوالي 18-30 شهرًا من العمر.
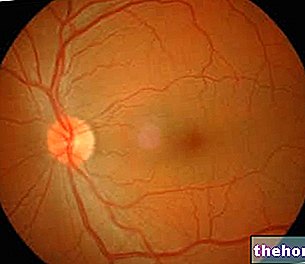
يتم تحديد التشخيص السريري للورم الأرومي الشبكي عن طريق فحص قاع العين ، وقد يظهر الورم ، حسب الموقع ، أثناء الفحص البسيط للعين ، من خلال تنظير العين غير المباشر. يمكن استخدام تقنيات التصوير لتأكيد التشخيص ، وتحديد مرحلة الورم (مكان وجوده ، ومدى انتشاره ، وما إذا كان يؤثر على وظائف الأعضاء الأخرى في الجسم ، وما إلى ذلك) وتحديد ما إذا كان العلاج فعالاً . قد تشمل الفحوصات الموجات فوق الصوتية والتصوير المقطعي المحوسب (CT) والتصوير بالرنين المغناطيسي (MRI).
التشخيص الجزيئي الجيني ممكن من خلال تحديد الطفرة في الجين RB1. يستخدم التحليل الوراثي الخلوي (أي الكروموسومات) للخلايا الليمفاوية في الدم المحيطي للكشف عن عمليات الحذف أو إعادة الترتيب التي تنطوي على الكروموسوم 13 (13q14.1-q14.2) .
العلاجات
في حالة الورم الأرومي الشبكي ، يمكن استخدام العديد من خيارات العلاج.
أهداف العلاج هي:
- القضاء على الورم وإنقاذ حياة المريض.
- احفظ العين إن أمكن ؛
- الحفاظ على الرؤية قدر الإمكان ؛
- تجنب تطور أنواع السرطان الأخرى ، والتي يمكن أن تنتج أيضًا عن العلاج ، خاصةً عند الأطفال المصابين بالورم الأرومي الشبكي الوراثي.
يعتمد التشخيص (احتمال الشفاء) وخيارات العلاج على العوامل التالية:
- مرحلة الورم
- عمر المريض وحالته الصحية العامة ؛
- موقع وحجم وعدد بؤر الورم ؛
- انتشار السرطان في مناطق أخرى بجانب مقلة العين
- ما مدى احتمالية الحفاظ على هذه الرؤية في عين واحدة أو كلتيهما.
يتم تشخيص معظم حالات الورم الأرومي الشبكي مبكرًا ومعالجتها بنجاح ، قبل أن ينتشر السرطان خارج مقلة العين ، مما يؤدي إلى معدل شفاء يزيد عن 90٪.